Bio Design
20th of September - George Church (Harvard)
Recordings
Presentations and slides
- 9:00 Megan Palmer - Review
- 10:00 George Church - Bio Design
- 11:45 Noah Jakimo - Homework Prep
Class assignments
DNA modification is fundamental to design in synthetic biology. For this week's assignment, you will learn to read DNA sequence files and see physical changes you will make to DNA molecules.
Lab Task: Verify restriction digest of a circular DNA plasmid by gel electrophoresis. Compare your experimental result with what you would predict from the plasmid map.
Theory Task: Choose a DNA sequence that interests you from nature or a source like the iGEM part registry. Provide a detailed workflow for isolating or acquiring a molecule with this specific sequence and an assay that confirms part of its content.
Hardware
| Function | Item |
|---|---|
| Incubating samples at 37C | Thermocycler, water bath, incubator, or body heat |
| Separating DNA by size | Gel electrophoresis box and power supply |
| Visualizing stained DNA | Blue-LED transilluminator |
| Preparing gel matrix | Microwave and balance |
Software
| Function | Item |
|---|---|
| Mapping out the pUC19 plasmid | DNA sequence viewer, such as Benchling, Snapgene, Geneious, ApE, GenBeans, or text editor |
Wetware
| Function | Item |
|---|---|
| Cutting DNA | Restriction enzyme (i.e. PvuII) and its buffer |
| Setting electrophoresis gel | Tris-acetate-EDTA (TAE) buffer and agarose |
Overview
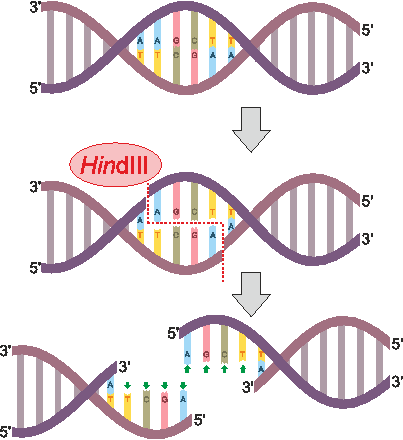
Tools to make specific cuts in DNA initiated discussion of “synthetic biology” as far back as the 1970s. The first of these genetic scissors were purified restriction enzymes, which are bacterial proteins that bind and break DNA at sequences they recognize to provide immunity to viruses. PvuII is an example of a restriction enzyme that recognizes CAGCTG and cuts (or “digests”) right down the middle (though restriction enzymes can also cut each strand in a way that leaves short overhangs, as seen for HindIII in the figure below). Our circular DNA plasmid pUC19 has >1 CAGCTG sites, which leaves multiple PvuII-cut fragments that can be separated into size-dependent bands by gel electrophoresis. This week we will cut DNA and compare the sizes of cut fragments to predicted outcomes from the sequence map of our plasmid.
Brief Protocol
1.In two separate tubes sized appropriately for your 37C source, mix the following
| Item | Vol to Rx |
|---|---|
| pUC19 ( 1ug / ul) | 2 ul |
| PvuII-HF (20 Units / ul) | 1 ul |
| Cutsmart Buffer (10x) | 3 ul |
| Deionized Water | 24 ul |
2.Incubate one reaction mix at 37C for 1 hr and move the other into a refrigerator (~4C)
3.Cast a 1% agarose TAE gel
- Add 1 g agarose to 100 mL 1x TAE (10 mL 10x TAE and 90 mL water) and swirl
- Microwave until agarose is fully dissolved (2-3 min) and before solution boils over
- Mix in 10000x DNA gel stain (10 uL for every 100 mL gel)
- Cast the gel in a tray with a well-forming comb inserted and wait ~1 hr to set (solidify)
4.Load gel with completed digests
- Orient solid gel so wells line up on the negative electrode side of the electrophoresis cell ("box")
- Fill gel box with 1x TAE until the gel is submerged and remove comb
- Mix samples with 6x DNA loading dye (6 ul dye for every 30 ul sample)
- Pipette DNA ladder into the first well, an uncut plasmid negative control into the second well, the 37C-incubated PvuII-cut sample into the third well, and the 4C-refrigerated sample into the fourth well.
5.Run for ~1 hr at 120V or until the dye front has traveled ¼ of the gel’s length
6.Image DNA separation through an orange filter over a blue-LED transilluminator.